
最新发布第2页

单细胞测序(三):寻找细胞标记
一、亚群注释 明确细胞类型。 1.识别每个类群的所有标记物; 2.识别每个类群的保留标记; 3.特定类群的标记识别。 1. 总体代码 markers <- FindAllMarkers(object = scRNA_harmony, test.use...

单细胞分析(一):矩阵数据的处理
一、总体代码 Seurat包官方文档https://satijalab.org/seurat/articles/pbmc3k_tutorial #加载所需R包 library(Seurat) library(tidyverse) library(dplyr) library(patchwork) library(ggplot2...

颅脑CT基础:CT断层解剖结构
一、颅CT基础 1.三种常用头颅扫描基线 (1)常规:横轴位(眦耳线),层厚8~10 mm (2)横轴位(眦耳线):上眶耳线(眦耳线向后倾斜20°) (3)鞍区扫描等 2.密度分类 低密度(黑-透光):空...

Linux系统通过密钥ssh登陆配置方法
一、制作密钥对 1.打开windows或者Mac OS中的powershell,输入 #生成密钥对 ssh-keygen -t RSA -b 2048 -C "your@domain.com" #-t 后表示ssh的密钥类型,常用的有:rsa、ed25519、dss...

文献学习2:创伤性脑损伤导致由小胶质细胞介导的慢性皮质炎症和神经元功能障碍
文献学习 Title:Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia DOI: https://doi.org/10.1523/JNEUROSCI.2469-20.2020 题目...

单细胞测序(四):软件注释
一、singleR 1. 总体代码 #第二种方法用SingleR鉴定细胞类型 ###下载好数据库后,把ref_Human_all.Rdata加载到环境中,这样算是对数据库的加载,就可以按照singler的算法来对细胞亚群进行定义了...
Hello, world!
疯狂肝了两天,此博客终于稳定下来。现在也终于可以有些时间写下第一篇文章,纪念一下这几天的付出。 一、建站灵感 说起建站的理由,其实也很偶然。前几天我在接收outlook邮件的时候,突然发现o...
python常见问题解决汇总(持续更新)
1.conda安装包提示Solving environment: unsuccessful initial attempt using frozen solve. Retrying with flexible solve 使用 Conda 安装包时遇到 “Solving environment: unsuccessful init...

WordPress跨服务器迁移最简单的手段
最近把博客建好了以后,一直在考虑如果有一天换服务器怎么办。于是就想把wordpress迁移流程走一遍,以便以后根据需要进行迁移。在这个过程中,又走了很多弯路,费了很多时间。也有一个好处,就...
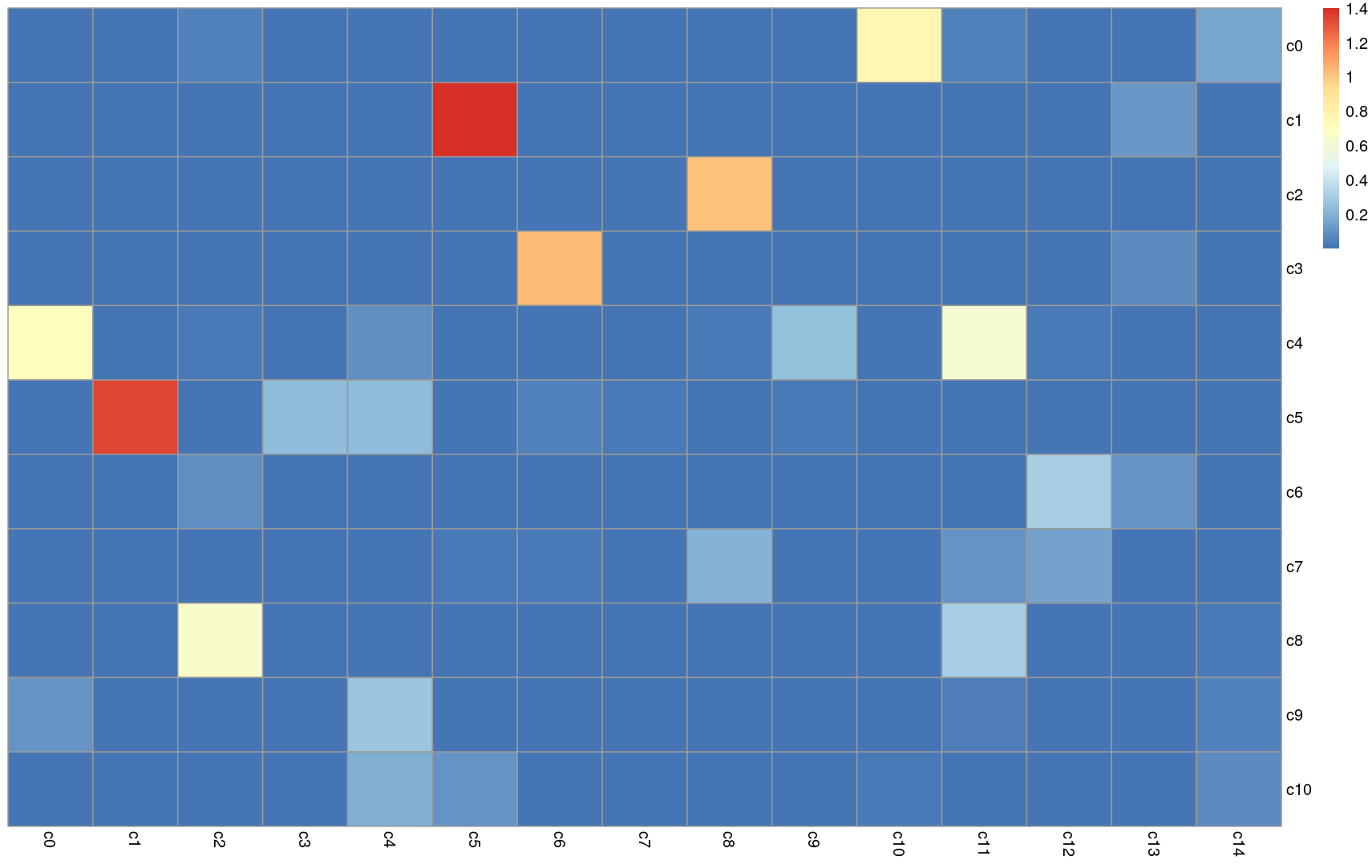
单细胞学习(五):通过聚类角度判断哪些cluster可能属于同一细胞类型
1.总体代码 ##清空环境 rm(list=ls()) #设置工作路径 ###加载所需要的包 library(Seurat) library(tidyverse) library(dplyr) library(patchwork) x=list.files() dir = c('BC2/', 'BC21/') nam...